Wetlab colonies - *Symbiodinium* qPCR
Running qPCR assays to see which Symbiodinium are in the colonies being held in the wetlab tanks.
Plate setup
MasterMix(n=48, assays=list("CD"))| GTMM | CF | CR | CP | DF | DR | DP | H20 |
|---|---|---|---|---|---|---|---|
| 480 | 48 | 48 | 48 | 48 | 48 | 48 | 96 |
# Get sample names
knitr::knit("../2017-02-15-sampling-intact-colonies-in-wetlab/2017-02-15-sampling.csv", quiet=T)[1] “2017-02-15-sampling.txt”
df <- read.csv("2017-02-15-sampling.txt")
colonies <- df$Tag..
colonies <- colonies[c(1:10,18,12,11,15,14,16,17,19,13)]
samples <- paste(rep(colonies, each=3), rep(c(1,2,3), len=length(colonies)*3), sep="-")
# Set up plate 1
plate1.samples <- c(samples[1:42], "Mc16-47","Mc14-78","Ss+", "NEC1", "NEC2", "NTC")
plate1 <- qPCRlayout(samples=plate1.samples, targets="CD")
knitr::kable(plate1)| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | CD / 946-1 | CD / 946-1 | CD / 947-3 | CD / 947-3 | CD / 943-2 | CD / 943-2 | CD / 950-1 | CD / 950-1 | CD / 933-3 | CD / 933-3 | CD / 940-2 | CD / 940-2 |
| B | CD / 946-2 | CD / 946-2 | CD / 944-1 | CD / 944-1 | CD / 943-3 | CD / 943-3 | CD / 950-2 | CD / 950-2 | CD / 938-1 | CD / 938-1 | CD / 940-3 | CD / 940-3 |
| C | CD / 946-3 | CD / 946-3 | CD / 944-2 | CD / 944-2 | CD / 949-1 | CD / 949-1 | CD / 950-3 | CD / 950-3 | CD / 938-2 | CD / 938-2 | CD / Mc16-47 | CD / Mc16-47 |
| D | CD / 945-1 | CD / 945-1 | CD / 944-3 | CD / 944-3 | CD / 949-2 | CD / 949-2 | CD / 936-1 | CD / 936-1 | CD / 938-3 | CD / 938-3 | CD / Mc14-78 | CD / Mc14-78 |
| E | CD / 945-2 | CD / 945-2 | CD / 948-1 | CD / 948-1 | CD / 949-3 | CD / 949-3 | CD / 936-2 | CD / 936-2 | CD / 937-1 | CD / 937-1 | CD / Ss+ | CD / Ss+ |
| F | CD / 945-3 | CD / 945-3 | CD / 948-2 | CD / 948-2 | CD / 942-1 | CD / 942-1 | CD / 936-3 | CD / 936-3 | CD / 937-2 | CD / 937-2 | CD / NEC1 | CD / NEC1 |
| G | CD / 947-1 | CD / 947-1 | CD / 948-3 | CD / 948-3 | CD / 942-2 | CD / 942-2 | CD / 933-1 | CD / 933-1 | CD / 937-3 | CD / 937-3 | CD / NEC2 | CD / NEC2 |
| H | CD / 947-2 | CD / 947-2 | CD / 943-1 | CD / 943-1 | CD / 942-3 | CD / 942-3 | CD / 933-2 | CD / 933-2 | CD / 940-1 | CD / 940-1 | CD / NTC | CD / NTC |
# Set up plate 2
plate2.samples <- c(samples[43:57], "NEC3", "NTC", "Mc16-47", "Mc14-78")
plate2 <- qPCRlayout(samples=plate2.samples, targets="CD")
knitr::kable(plate2)| 1 | 2 | 3 | 4 | 5 | 6 | |
|---|---|---|---|---|---|---|
| A | CD / 939-1 | CD / 939-1 | CD / 934-3 | CD / 934-3 | CD / NTC | CD / NTC |
| B | CD / 939-2 | CD / 939-2 | CD / 932-1 | CD / 932-1 | CD / Mc16-47 | CD / Mc16-47 |
| C | CD / 939-3 | CD / 939-3 | CD / 932-2 | CD / 932-2 | CD / Mc14-78 | CD / Mc14-78 |
| D | CD / 935-1 | CD / 935-1 | CD / 932-3 | CD / 932-3 | CD / 939-1 | CD / 939-1 |
| E | CD / 935-2 | CD / 935-2 | CD / 941-1 | CD / 941-1 | CD / 939-2 | CD / 939-2 |
| F | CD / 935-3 | CD / 935-3 | CD / 941-2 | CD / 941-2 | CD / 939-3 | CD / 939-3 |
| G | CD / 934-1 | CD / 934-1 | CD / 941-3 | CD / 941-3 | CD / 935-1 | CD / 935-1 |
| H | CD / 934-2 | CD / 934-2 | CD / NEC3 | CD / NEC3 | CD / 935-2 | CD / 935-2 |
Results
Raw data files
*.eds
*.txt
Analysis
# Import steponeR function
source_url("https://raw.githubusercontent.com/jrcunning/steponeR/master/steponeR.R")## SHA-1 hash of file is 5d640f385ee8e972e6c6e54b60abb88689e04046# List results files
plates <- list.files(pattern = "*_data.txt", full.names = T)
# Import data using steponeR
q <- steponeR(files=plates, delim="\t", target.ratios="C.D",
fluor.norm=list(C=2,D=0))$result## Loading required package: plyr## Loading required package: reshape2# QC data (remove amps with only one technical rep)
q$C.D <- ifelse(q$C.reps < 2, ifelse(q$D.reps < 2, NA, -Inf), ifelse(q$D.reps < 2, Inf, q$C.D))
# Calculate proportion D in each sample (=D/(C+D))
q$propD <- ifelse(is.finite(q$C.D), 1/(q$C.D+1),
ifelse(q$C.D > 0, 0, 1))
# Merge with colony metadata
q$Colony <- substr(q$Sample.Name, 1, 3)
df$Colony <- df$Tag..
res <- merge(df, q[,c("Colony", "Sample.Name", "propD")])[,c(1,3,7,8)]
res$Species <- as.character(res$Species)
res$Species[grep(pattern="Ofav", res$Species)] <- "Orbicella"
res$Colony <- factor(res$Colony)
# Plot results
library(lattice)
par(mfrow=c(1,3))
with(res[res$Species=="Ssid",], xyplot(propD ~ Colony, ylim=c(0,1), cex=2, main="S. siderea"))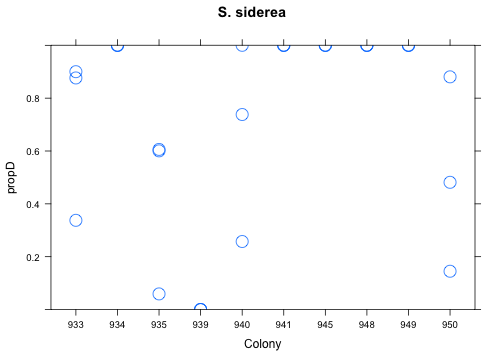
with(res[res$Species=="Mcav",], xyplot(propD ~ Colony, ylim=c(0,1), cex=2, main="M. cavernosa"))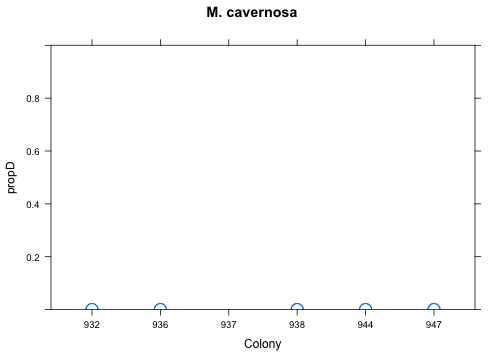
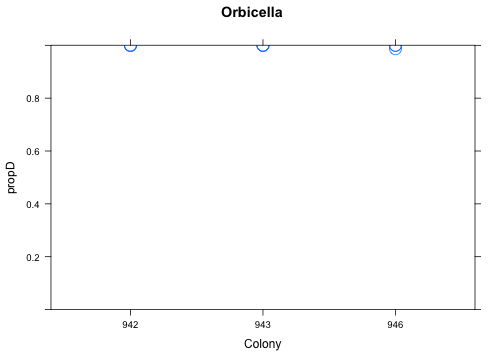
with(res[res$Species=="Orbicella",], xyplot(propD ~ Colony, ylim=c(0,1), cex=2, main="Orbicella"))