ITS2 clustering
I am thinking about how to improve OTU clustering of Symbiodinium ITS2 sequences in the face of intragenomic variation and the presence of identical sequence variants in multiple different types of Symbiodinium. For example, Figure 4 in Arif et al. (2014) shows ITS2 sequences recovered from four different members of Symbiodinium clade C.
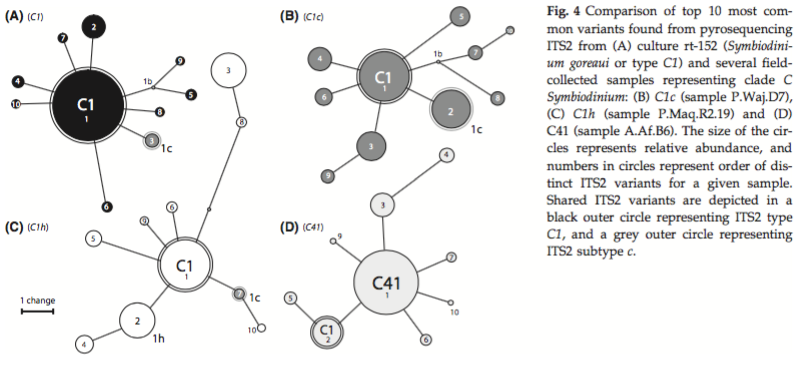
These four different types all contain the C1 sequence variant at different relative proportions within the rDNA array. All of these clade C ITS2 sequence variants clustered into a single 97%-identity OTU, which was subsequently named C1 because the most abundant sequence in the OTU was the C1 sequence. However, panel D represents a symbiont type whose dominant sequence variant is not C1, but C41. Because sequences from all samples were pooled and clustered together, these sequences were subsumed into the C1 OTU because C1 is the most abundant variant among all pooled sequences. However, if clustering was performed for each sample individually, C41 would be the most abundant sequence variant in panel D and a separate OTU would be formed with C41 as the representative sequence. However, panels A, B, and C, which may represent distinct clade C types C1, C1c, and C1h, would all still collapse into a single OTU with C1 as the representative sequence, because C1 is most abundant within each of these samples as well. Therefore, within-sample clustering would help resolve some, but not all, distinct OTUs that would otherwise be collapsed together when clustering is performed on pooled sequences.